When you pick up a prescription at the pharmacy and it’s not the brand-name drug you expected, that’s not a mistake. It’s the result of one of the most important, yet often overlooked, systems in American healthcare: the FDA’s approval of generic drugs. Around 9 out of 10 prescriptions filled in the U.S. are for generics. But how does the FDA make sure these cheaper versions are just as safe and effective as the expensive brand-name drugs? The answer lies in a 40-year-old law, a detailed scientific process, and a regulatory pipeline that’s been fine-tuned to deliver results - without cutting corners.
The Legal Backbone: The Hatch-Waxman Act of 1984
Before 1984, generic drug manufacturers faced a near-impossible hurdle. To get approval, they had to repeat every single clinical trial that the original drugmaker had done - even if the active ingredient was identical. That meant spending hundreds of millions of dollars and waiting over a decade just to enter the market. Most couldn’t afford it. The result? Few generics, high prices, and limited access. That changed with the Drug Price Competition and Patent Term Restoration Act - better known as the Hatch-Waxman Act. Signed into law on September 24, 1984, this legislation created the Abbreviated New Drug Application (a regulatory pathway that allows generic drug manufacturers to seek approval without repeating full clinical trials, relying instead on the FDA’s prior findings on the brand-name drug) (ANDA). This wasn’t a shortcut - it was a smart one. It let generics skip expensive animal and human trials by using the FDA’s own data on the original drug. The law also gave brand-name companies a limited window of market exclusivity to recoup R&D costs, balancing innovation with competition. Today, the ANDA pathway is the backbone of the U.S. generic drug system. It’s not just a policy - it’s the reason why a 30-day supply of metformin costs $4 instead of $150.What the FDA Actually Checks: The 5 Requirements for Approval
The FDA doesn’t approve generics based on trust. It demands proof. For a generic drug to get the green light, it must meet five strict criteria:- Same active ingredient - No surprises here. The generic must contain the exact same active chemical compound as the brand-name drug. No extra, no less.
- Identical strength, dosage form, and route - If the brand is a 500mg tablet taken orally, the generic must be too. No capsules, no injections, no different doses.
- Same use indications - It must treat the exact same conditions. A generic for high blood pressure can’t suddenly be marketed for diabetes.
- Bioequivalence - This is the golden standard. The generic must deliver the same amount of drug into the bloodstream at the same rate as the original. This is tested in 24-36 healthy volunteers using blood samples taken over time. The FDA requires the generic’s absorption to fall within 80-125% of the brand’s - a narrow window that ensures consistent therapeutic effect.
- Identical manufacturing standards - The factory where the generic is made must follow the same Good Manufacturing Practices (GMP) as the brand. The FDA inspects these facilities - often unannounced - and shuts down non-compliant sites.
The ANDA Submission: What’s in the File?
Submitting an ANDA isn’t a simple form. It’s a 500-1,000 page dossier packed with technical data. Here’s what’s required:- Chemistry, Manufacturing, and Controls (CMC) - Detailed info on how the drug is made, including raw materials, equipment, and quality control steps.
- Facility details - Every manufacturing, packaging, and testing site must be listed and approved.
- Proposed labeling - The package insert must match the brand’s in content, though it can omit the brand name.
- Bioequivalence study results - Full data from the clinical trials, including statistical analysis and subject demographics.
- Patent and exclusivity certifications - The applicant must certify whether the brand’s patents are expired, invalid, or will be challenged.
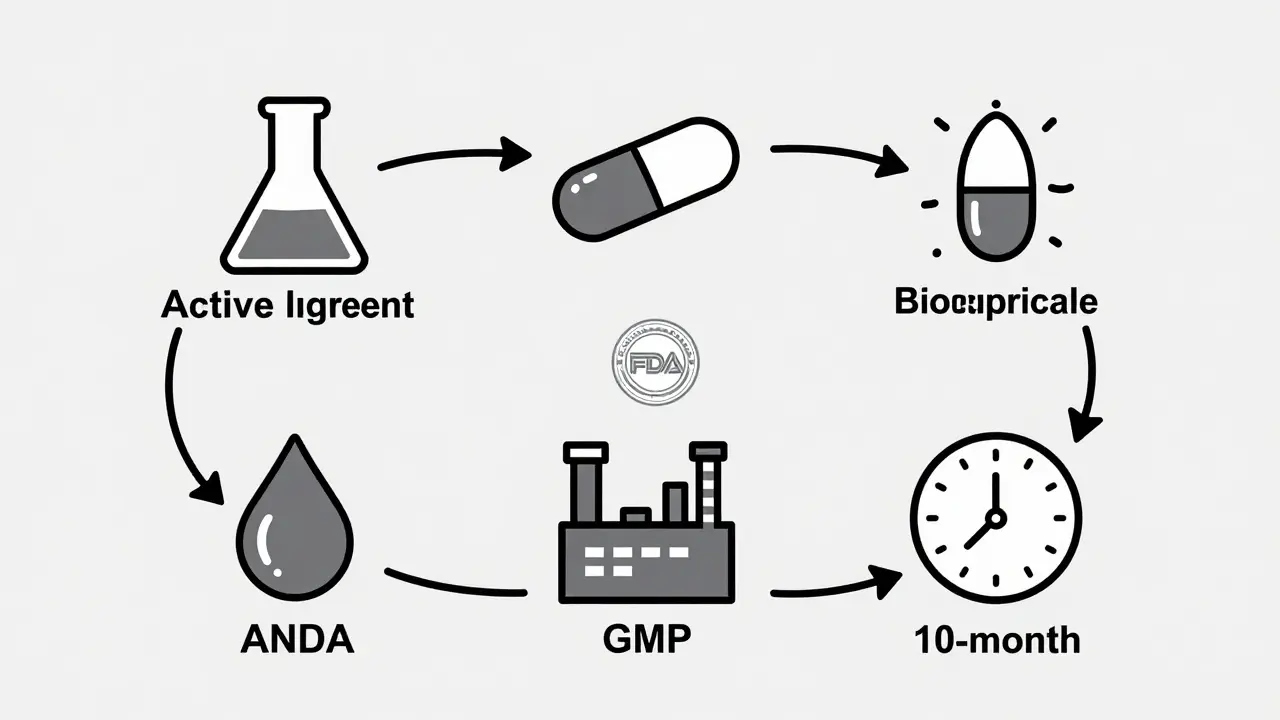
Review Timeline: From Submission to Approval
The timeline isn’t what it used to be. Before 2012, reviews could drag on for years. Today, thanks to the Generic Drug User Fee Amendments (a funding system that lets the FDA collect fees from generic manufacturers to improve review efficiency and set clear timelines) (GDUFA), the clock is ticking.- Standard ANDAs - Reviewed within 10 months of submission.
- Priority ANDAs - For drugs in shortage or first generics - reviewed in 8 months.
Costs and Competition: Why Generics Are So Much Cheaper
A brand-name drug can cost over $2.6 billion to develop. That includes failed drugs, clinical trials, marketing, and patent protection. A generic? Typically $1-5 million. Why? Because it doesn’t repeat the original research. It just proves it works the same way. The result? Generics cost 80-85% less than their brand-name counterparts. That’s not inflation - that’s competition. In 2022, the U.S. generic drug market was worth $125 billion. Teva, Sandoz, Mylan (now Viatris), and Amneal dominate the space, but hundreds of smaller firms are pushing into complex generics - like inhalers, injectables, and topical creams - where the science gets harder. The FDA’s Drug Competition Action Plan (a strategy to remove barriers to generic entry, especially for drugs with limited competition) targets exactly those areas. For example, the approval of the first generic version of Vivitrol (naltrexone extended-release injection) in 2023 was a big deal - not just because it was cheaper, but because it helped expand access to addiction treatment during the opioid crisis.
Where the System Gets Tricky: Complex Generics
Not all drugs are created equal. A simple tablet? Easy. An inhaler that delivers a precise dose to the lungs? Hard. A topical cream that penetrates skin layers consistently? Even harder. For these complex generics (drugs with intricate delivery systems that require more than bioequivalence testing to prove equivalence), the standard ANDA isn’t enough. The FDA has launched the Complex Generic Drug Product Development Resources (an initiative to help manufacturers navigate the science behind complex generics) to help. These require additional studies - sometimes in patients, sometimes with specialized equipment - to prove they’re truly equivalent. That’s why some generics still take years to appear. The science is harder. The data is bulkier. The FDA’s review teams are still learning how to evaluate them.The Bigger Picture: Why This Matters
This isn’t just about drug prices. It’s about access. A diabetic who can’t afford insulin. A cancer patient who can’t get their maintenance chemo. A veteran who needs daily medication but lives on a fixed income. Generics make all of those scenarios manageable. The FDA’s system isn’t perfect. Patent thickets, legal delays, and manufacturing bottlenecks still happen. But the framework - rooted in science, not speculation - works. It’s why 90% of prescriptions are filled with generics. It’s why the U.S. spends less on drugs than most other developed countries. And now, there’s a new twist. In October 2025, the FDA announced a pilot program that prioritizes ANDA reviews for companies that test and manufacture their drugs in the U.S. It’s not just about speed - it’s about building a domestic supply chain. After years of relying on overseas factories, the U.S. is betting on homegrown generics.What’s Next?
The future of generics isn’t just about more pills. It’s about smarter approvals. More transparency. Better tools for complex products. And more incentives to make them here. The system isn’t going away. It’s evolving. And as long as the FDA keeps its standards high - and its deadlines tight - generics will keep saving lives and saving money.Are generic drugs as safe as brand-name drugs?
Yes. The FDA requires generics to have the same active ingredient, strength, dosage form, and route of administration as the brand-name drug. They must also prove bioequivalence - meaning they deliver the same amount of drug into the bloodstream at the same rate. Every manufacturing facility is inspected under the same strict standards. If a generic passes review, it’s just as safe and effective.
Why do some generics look different from the brand-name drug?
The active ingredient must be identical, but inactive ingredients - like fillers, dyes, and coatings - can differ. These don’t affect how the drug works, but they can change the color, shape, or taste. That’s why your generic pill might look different. It’s still the same medicine.
Can a generic drug be approved before the brand-name patent expires?
Yes - but only if the generic manufacturer challenges the patent. This is called a Paragraph IV certification. If the brand-name company sues for infringement, the FDA can’t approve the generic for up to 30 months. If the court rules in favor of the generic, approval can happen earlier. Many first generics are approved this way.
How does the FDA ensure quality in generic manufacturing?
The FDA inspects every manufacturing site - domestic and foreign - using the same standards as brand-name facilities. Facilities are inspected every 2-3 years, often unannounced. If a site fails inspection, the FDA can refuse to approve the drug or even halt production. Quality is non-negotiable.
What happens if a generic drug fails bioequivalence testing?
The application is rejected. The manufacturer must fix the issue - whether it’s the formulation, manufacturing process, or testing method - and resubmit. Many companies go through multiple rounds before approval. The FDA doesn’t approve a generic just because it’s cheaper. It approves it only when it’s proven to work the same way.
For patients, doctors, and pharmacists, the system works because it’s built on proof - not promises. That’s why you can trust a generic.


Lillian Knezek
February 24, 2026 AT 17:43Maranda Najar
February 26, 2026 AT 05:40Christopher Brown
February 27, 2026 AT 02:14Alfred Noble
February 28, 2026 AT 13:03Matthew Brooker
March 1, 2026 AT 08:23Emily Wolff
March 2, 2026 AT 03:38David McKie
March 3, 2026 AT 07:49Southern Indiana Paleontology Institute
March 5, 2026 AT 00:42